Kass, Jason, Ruben Artero, and Mary K. Baylies. 2000. Non-radioactive electrophoretic mobility shift assay using digoxigenin-ddUTP labeled probes. Dros. Inf. Serv. 83: 185-188.
|
|
|
|||
|
|
||||
Non-radioactive
electrophoretic mobility shift assay using digoxigenin-ddUTP labeled probes.
Kass,
Jason, Ruben Artero, and Mary K. Baylies. Program in Molecular Biology, Sloan Kettering
Institute, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York,
New York 10021
Electrophoretic
mobility shift assay (EMSA) is a powerful technique used to quantitatively
analyze sequence specific DNA binding proteins. Traditionally a radiolabeled DNA probe is added to a candidate
protein and then the mixture is separated on an acrylamide gel. When a DNA-protein complex forms, it is
retarded by the gel and appears “shifted” in comparison to the
mobility of the free probe. This
apparent complex can then be challenged with an unlabeled probe of the same
or different sequence to establish specificity. Since this method uses a radiolabeled probe, it not only requires
the safety concerns associated with proper storage, usage and disposal of
radioactivity, but also limits the lifespan of the probe. Here we present the use of Digoxigenin
labeled probes as an alternative to radioactivity in EMSA. In this assay the shift reaction is resolved
on an acrylamide gel and then transferred to a nylon membrane for detection
using an antibody against Digoxigenin. It offers the advantages of high resolution and comparable
sensitivity of 32P labeled probes. In addition this method is well suited for teaching students
new biochemical techniques.
To demonstrate the effectiveness of this
method we tested the ability for Twist, a basic Helix-Loop-Helix transcription
factor, to bind a 20 bp double-stranded DNA fragment from the rhomboid promoter of D. melanogaster.
This fragment contains a canonical E-box (CATATG) to which Twist has
been previously shown to bind (Ip et al., 1992). Twist
binds DNA by forming homodimers with itself or heterodimers with other bHLH
proteins to either activate or repress myogenesis (Castañon et al., manuscript submitted).
Labeling Probe with Digoxigenin: 1 nmol
of each oligonucleotide (5´-GATCCCTCGCATATGTTGAA-3´ Top, 5´-GATCTTCAACATATGCGAGG-3´
Bottom) was annealed by heating at 95ºC for ten minutes and slowly cooled
to room temperature in a buffer of Tris (10 mM, pH 8), EDTA (1 mM, pH 8) and
NaCl (100 mM). The double-stranded oligonucleotide was
then labeled using the reagents provided in the DIG Oligonucleotide 3´-End
Labeling Kit (Roche Cat. No. 1 362 372).
The labeling protocol was modified only in the incubation and precipitation
times. 100 pmol of the fragment
was added (on ice) to 4 ml 5´ labeling buffer, 4 ml, 25 mM CoCl2, 1 ml 1 mM DIG-11-ddUTP and 2.5 U Terminal Transferase
in a final volume of 20 ml and incubated at 37ºC for 1 hour. The DNA was then precipitated with 2 ml 4 M LiCl and 60 ml 100% ice-cold ethanol and incubated at
–70ºC for 2 hours. The
precipitated DNA was then pelleted by centrifugation for 15 minutes at 4ºC,
13,000 ´ g, washed
once with 100 ml ice-cold
70% ethanol and recentrifuged for an additional 15 minutes. After air drying the pellets, the DNA
was resuspended in ddH2O to a final concentration of 2.5 pmol/ml.
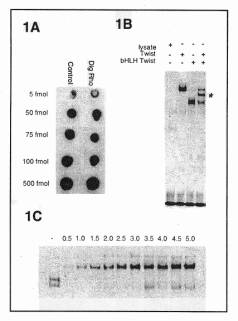
Generating a dot blot, as shown in Figure 1A, tested the efficiency
of the labeling reaction. Serial
dilutions were spotted on a nylon membrane and detected as described below.
The resulting intensity was then directly compared to the control labeled
oligonucleotide provided with the labeling kit.
The probe was then stored at –20ºC.
Shift Reaction: Full-length twist, bHLH twist and tethered twist-twist DNA constructs were used as templates in a TnT Coupled Reticulocyte Lysate System (Promega Cat. No. L4610, Castañon et al., manuscript submitted). The bHLH twist construct is a truncated form missing amino acids 141-331 yet retains both the dimerization and DNA binding domains. The twist-twist tethered dimer has two full length twist monomers connected by a serine and glycine flexible linker. In vitro translations were performed using 150 ng of input DNA in a 7.5 ml reaction containing 3.75 ml of rabbit reticulocyte lysate. The protein products were incubated in 2 mM DTT at 37ºC for 10 minutes (Marcus, 2000). After a 5-minute equilibration to 25ºC, 5 ml of the protein products were added to 50 fmol of the Digoxigenin labeled probe. The 25 ml reaction mixture contained 1 mg poly d(I-C) (Roche Cat. No. 108 812), 2 mM MgCl2, 25 mM HEPES pH 7.5, 100 mM NaCl, 0.1% Igepal CA-630 (Sigma Cat. No. I-3021), 12% glycerol (v/v). The mixture was electrophoresed at 10 volts/cm through a 0.25´ TBE – 5 % polyacrylamide gel (acrylamide-bisacrylamide, 29:1 - 3.3 % C). The 15 cm ´ 15 cm gel was then transferred to a positively charged nylon membrane (Roche Cat. No. 1 209 272) using a semi-dry transfer cell (Bio Rad) under a constant amperage of 0.56 mA (25 V max) for 15 minutes. The DNA was then cross-linked to the membrane using a UV stratalinker (Stratagene).
Detection: 10´
blocking reagent (Roche Cat. No. 1 096 176) was prepared in a maleic acid
buffer [0.1 M maleic acid (pH 7.5), 0.15 M NaCl] according to the manufacturer.
The nylon membrane was blocked for one hour at room temperature in
2´ blocking reagent and
then incubated with Anti-Digoxigenin coupled to Alkaline Phosphatase (Roche
Cat. No. 1 093 274) diluted 1:20,000 for 30 minutes. The membrane was washed twice for 20 minutes each with 200
ml of maleic acid buffer with 0.3% Tween 20 added. After a 5 minute equilibration to pH 9.5 in 50 ml of the detection
buffer (100 mM Tris-HCl, pH 9.5, 100 mM NaCl) the membrane was carefully placed
on a plastic sheet protector. The detection substrate, CDP-Star (Roche Cat. No. 1 685 627)
was diluted 1:100 in detection buffer.
2 ml of diluted substrate was used to cover the membrane. As the CDP-Star can precipitate when stored
at 4ºC, it was first warmed to room temperature and briefly vortexed.
To ensure low background, care was taken to add the substrate dropwise
around the edges of the membrane before covering the entire membrane by tilting. After incubating the membrane for 5 minutes,
it was briefly blotted on Whatman paper and placed between two pieces of plastic.
The chemiluminescence required 15 minutes to peak and the membrane
was then exposed for 5 minutes using Hyperfilm-MP (Amersham Cat. No. RPN1675H).
Results
and Discussion
As demonstrated in Figure 1B, both the full length Twist (Lane 2) and
truncated bHLH Twist (Lane 3).
independently
form strong homodimers, shown by their intense, sharp bands. When co-translated, a mixture of three
distinct complexes can form. These
include the individual homodimers and a Twist/bHLH Twist heterodimer of intermediate
mobility. Lane 4 shows that all
three complexes can be resolved very clearly using this technique.
Digoxigenin-labeled shifts provide excellent resolution that is useful
when identifying distinct species that migrate closely
Non-radioactive EMSA has several advantages over the traditional radioactive
approach. Being non-radioactive, the labeled probes
are safer to handle in the laboratory. Although the labeling reaction buffer contains Potassium cacodylate,
a toxic chemical, once labeled the DNA probes require no additional handling
precautions or disposal methods unlike 32P waste. Digoxigenin labeled probes are also more
stable than radioactive probes. Whereas
32P labeled probes have a half-life of two weeks, Digoxigenin labeled
probes are stable for much longer periods of time. We have recently used probes that were
labeled over a year earlier.
This technique is also competitive in terms of sensitivity, time commitment
and cost. The range of input
DNA (50 fmol, 0.6ng) and in vitro translated protein (5ml) is consistent with radioactive
shifts of other Helix-Loop-Helix family members (Benezra et al., 1990 and Murre et al.,
1989). As seen in Figure 1C the
Twist-Twist tethered homodimer can be detected using as little as 1.0 ml of
in vitro translated product. Similar
results were achieved with a number of other bHLH proteins. This titration illustrates the range of
|
|
Figure
1. Digoxigenin labeled
oligonucleotide from the rhomboid promoter used in EMSA. (A) Testing efficiency of the labeling
reaction using a dot blot. 1
ml
of 5 serial dilutions were spotted next to dilutions of a control labeled
oligonucleotide. (B) In
vitro translated products of Twist and bHLH Twist were assayed for binding
to the rhomboid promoter in an EMSA using Digoxigenin labeled oligonucleotide
as probe. 5 ml
of the following products were added to each lane: 1, unprogrammed lysate; 2, Twist; 3,
bHLH Twist; 4, co-translated
Twist and bHLH Twist. Asterisk
shows heterodimer. Free
probe is at bottom of gel. (C)
The linked dimer Twist-Twist was added in 0.5 ml increments to show the
range of sensitivity of the non-radioactive EMSA. Lane 1 contains 3 ml of unprogrammed lysate.
The free Digoxigenin probe was in excess for each lane. |
We have found non-radioactive EMSA to be a sensitive, efficient, safe and durable technique. Most importantly the shift complexes give excellent resolution and are easily identifiable. This robust technique can also serve as an excellent teaching tool for undergraduate laboratory classes.
References: Benezra, et al., 1990, Cell
61: 49-59; Castañon, et
al. (manuscript submitted).
Dimerization partners determine the activity of the Twist bHLH protein
during Drosophila mesoderm development;
Ip, et al., 1992, Genes
Dev. 9: 1728-1739; Murre, et
al., 1989, Cell 58: 537-544.