
Webster, Brian. 2001. A quantitative study of the ability of cryopreservation to stabilize nucleic acids in Drosophila tissue specimens. Dros. Inf. Serv. 84:157-165.
|
|
|
|||
|
|
||||
A quantitative study of the ability of cryopreservation to stabilize nucleic acids in Drosophila tissue specimens.
Webster, Brian.* Ambrose Monell Collection for Molecular and Microbial Research, American Museum of Natural History, Central Park West at 79th St, New York, NY, 10024. *Current Address: 012 Baker North Hall, Cornell University, Ithaca, NY, 14853. Email: brw26@cornell.edu
Abstract
A quantitative
PCR (QPCR) study was undertaken to assess the quality of DNA isolated from
individual Drosophila melanogaster
specimens. The control samples were frozen cryogenically
using a number of different methods, involving freezing the samples in the
following cryoprotective media: 1.5 M DMSO, 1.5 M glycerol, 1.2 M ethylene
glycol, 100% ethanol, and 70% (v/v) ethanol, as well as “dry”
freezing, that is, with no added cryoprotectant. A number of different freezing and thawing
protocols were also explored for samples frozen in each cryoprotectant: rapid
(freezing) / slow (thawing), rapid / rapid, slow / rapid, and slow / slow.
All samples in the study were then stored at -159°C for 1-3 weeks, then assayed
for DNA quality using QPCR amplification of a ~800 bp fragment in the mitochondrial
cytochrome oxidase II region. In addition, the QPCR test was performed
on control samples of D. melanogaster not subjected to equilibration in cryoprotectant or
freeze/thawing. The control samples
showed a significantly higher measured quality of DNA than any of the cryopreserved
samples (Mann-Whitney Rank Sum, p < 0.01), as determined by the ratio of
mass of amplified product to mass of template used in the PCR reaction.
It was found that the DNA quality as a whole of the slow freeze / rapid
thaw and the slow freeze / slow thaw methods was significantly higher than
that of the rapid freeze / rapid thaw and the rapid freeze / slow thaw methods,
respectively (p < 0.05). It
was also determined that slow freezing in no cryoprotectant, irregardless
of thawing protocol, resulted in significantly higher quality DNA than any
other cryopreservation protocol (p < 0.05).
A qualitative comparison was done using the PCR test on Drosophila
pseudoobscura stored in 70% ethanol at room temperature for 15 years.
No amplification was observed in two non-quantitative PCR reaction
tests performed on four D. pseudoobscura samples.
Introduction
The traditional focus in museum collections has been on preserving organisms or tissues for morphological studies to determine phylogeny. However, in the last decade it has become clear that genomics is perhaps as equally important a tool in phylogenetic studies as morphology. Ethanol preservation became a useful procedure in preserving tissues for molecular studies as ethanol likely inhibits proteins that degrade nucleic acids, such as DNases, while preserving the nucleic acids themselves (Flournoy et al., 1996) . However, ethanol is known to induce strand breaks and chromosomal aberrations (Tateno et al., 1998; Blasiak et al., 2000) ; additionally, long-term storage in ethanol allows significant DNA degradation (Dick et al., 1993; Barnes et al., 2000) . Storage at room temperature (even in ethanol) further permits occurrences of biochemical reactions and osmotic exchanges to continue.
Tissue storage at temperatures below the glass transition temperature of water (-139°C) is known stop essentially all chemical reactions from occurring (i.e., DNase degradation); a chemical reaction at -200°C occurs approximately 8 million times slower than the same reaction at 0°C (Grout and Morris 1987) . However, freezing is known to stress cells chemically by increasing solute concentration as ice crystallizes, causing cytoplasmic water loss and to stress cells mechanically by ice crystallization (Grout and Morris 1987) . Some prior studies using the following assays: terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) (Duru et al., 2001) , the comet assay (Steele et al., 2000) , an X-linked recessive lethal assay in cryopreserved embryos (Houle et al., 1997) , and multilocus minisatellite probe comparisons (Ross et al., 1990) , have not found a correlation between cryopreservation and DNA damage. However, this research is far from conclusive, as a separate comet assay analysis did find evidence of DNA damage (Belpaeme et al., 1998) . Other assays have been performed which also showed evidence of DNA damage in cryopreserved cells: terminal restriction fragment (TRF) analysis (Honda et al., 2001) , sister chromatid exchange (SCE) test (Bouquet et al., 1993) , and visual chromosome analysis (Shaw et al., 1991) . Therefore, there seems to be no clear consensus on whether cryopreservation causes genetic damage, significant or otherwise.
The polymerase chain reaction (PCR) is quickly becoming one of the most important tools in molecular research, especially in the phylogenetic studies performed in museums that might be interested in tissue cryopreservation as a means of banking genetic diversity. With the conflicting research on genetic damage as a result of cryopreservation and the dearth of quantitative research on the ability to perform PCR on cryopreserved tissues, we decided to perform a quantitative PCR analysis on the level of genetic damage as a result of cryopreservation. The PCR analysis compared the ability of the polymerase to amplify a specific segment of DNA from the unfrozen controls to the cryopreserved experimental samples.
In addition, we used the quantitative PCR assay to analyze the best protocol for cryopreserving tissues. Cryopreservation came about as a means of banking reproductive and transplantable tissues for later use. Since then, a number of chemicals (cryoprotectants) have been found to increase the viability of these tissues by protecting them from the stresses of freezing and thawing if the tissue was equilibrated in the cryoprotectant before freezing (Parkening et al., 1976; Kasai et al., 1981; Ingham et al., 1993; Fisher et al., 1996) . Additionally, the rate of cooling and thawing was found to affect viability after cryopreservation (Kasai et al., 1981; Grout and Morris 1987) . It has been shown, not surprisingly, that the same cryoprotectants and rates of freeze/thaw used to increase viability have also been found to affect the level of genetic damage as a result of cryopreservation (Shaw et al., 1991) .
We decided in
this study to use tissue samples applicable to museum research collections,
i.e., whole organisms (D. melanogaster and D. pseudoobscura). The
cryoprotective agents and the particular molarities, as well as rates of freezing
and thawing, were decided upon because of their popularity and widespread
use in cryopreservation and tissue preservation studies
(Parkening et al., 1976; Kasai
et al., 1981; Ingham et al., 1993; Dillon
et al., 1996; Fisher et al., 1996)
. Since the whole organism was to be preserved, and post-thaw
viability was not a concern with these tissues, the usual addition of serum
or protective buffer to the cryoprotective solutions was forgone. The DNA was extracted by an easily repeatable,
uniform method (Qiagen DNeasy tissue kit). The quantitative PCR assay was modified
slightly from a well-established method
(Ayala-Torres et al., 2000)
, to better fit the equipment at hand (see Methods for details).
Methods
Freezing and Thawing Procedures
A total of 24 freezing protocols were performed, using an aliquot of
20 Drosophila melanogaster individuals
for each protocol. The 24 protocols
were comprised of six cryoprotectants each frozen/thawed using four methods. The six cryoprotectants used were 1.50
M molecular grade dimethyl sulfoxide, 1.50 M enzyme grade glycerol, 1.20 M
enzyme grade ethylene glycol, 70% (v/v) ACS/USP grade ethanol, and 100% ethanol,
as well as a “dry” freezing aliquot with no cryoprotectant added.
The cryoprotectants were diluted to the correct molarity or concentration
in distilled water, not serum or buffer.
The four freeze/thaw protocols were: a rapid freeze/rapid thaw, rapid
freeze/slow thaw, slow freeze/rapid thaw, and slow freeze/slow thaw.
Rapid cooling
1200 mL of
cryoprotectant solution was added to a 2.0 mL cryogenic vial (Corning Inc.)
and the tubes were cooled to 0°C in an ice bath. The flies were anaesthetized by placing in a -20°C
freezer for 5-10 min, and 20 individuals were removed and placed in the cryogenic
vials. The vials were inverted
to immerse the flies in the cryoprotectant and the vials were then equilibrated
in the cryoprotectant at 0°C in an ice bath for
2 hours. After the equilibration
period was complete, the vials were immersed in liquid nitrogen. The vials were then removed from the liquid
nitrogen and placed in an XLC 1830HE vapor-phase freezer (MVE, Bloomington,
MN) at approximately -159°C. The vials were stored for 2-3 weeks before
removing from the freezer.
Slow cooling
600 mL of cryoprotectant solution was added to the 2.0 mL cryogenic vials, cooled to 0°C. The flies were placed in the vials, the vials were inverted, and the vials were equilibrated at 0°C for 90 min. After the 90 min equilibration period was complete, an additional 600 mL of cryoprotectant was added. The vials were equilibrated for a further 90 min at 0°C. The vials were then placed in a Cryo 1°C Freezing Container (Nalgene). The deviations from the protocol set for the container were that 70% (v/v) isopropyl alcohol was used instead of 100% isopropyl alcohol, and the container was placed in a -75°C freezer for 3 hours instead of Nalgene’s established protocol involving cooling at –70°C for 4 hours. After the cooling period was complete, the vials were removed from the Cryo container, immersed in liquid nitrogen, and placed in the XLC 1830HE vapor-phase freezer.
Rapid Thaw. The vials were removed from the freezer and placed in a 37°C water bath. The vials were equilibrated for 3 min, at which point one fly was removed and the vials were refrozen according to the original cooling method.
Slow Thaw. The vials were removed from the freezer and equilibrated in air at room temperature (~21°C) for 30 min. A single fly was removed and the vials were refrozen according to the original cooling method.
Controls. The protocol to assess the quality of DNA was also performed on Drosophila melanogaster not subjected to equilibration in a cryoprotectant or any freeze/thaw procedure. These controls were performed as a comparison of DNA from “fresh” tissue as opposed to DNA from tissue subjected to cryopreservation protocols. A total of four flies were used as control measurements, with four corresponding DNA extractions.
Ethanol-Preserved
Flies. A non-quantitative test was performed on
Drosophila pseudoobscura stored in
70% ethanol for 15 years. The
DNA quality assessment protocol was the same for these flies as for all others,
but the obvious difference in storage technique and duration negated the usefulness
of quantifying the quality of the DNA of these flies. This protocol was done simply as a method of comparison. A total of four flies were used, with
four corresponding DNA extractions.
DNA Quality Evaluation
DNA Extraction. Total genomic DNA was extracted from a single fly using a Qiagen DNeasy Tissue Kit. The protocol for the kit was followed, except for the following deviations. The flies were not ground in liquid nitrogen but simply homogenized in buffer ATL to prevent possible damage to DNA by freezing. Secondly, after lysis was complete, the sample was treated with 4 mL RNase A (100 mg/mL), and the final elution in buffer AE was only done using a single 50 mL elution, since the mass of a single fly is only ~0.3 mg.
Total Genomic DNA quantification. The DNA in the elution was quantified spectrophotometrically using a GeneQuant pro RNA/DNA Calculator (Amersham Pharmacia Biotech). 10 mL of the elution was removed and pipetted into an ultra micro volume cell for quantification. 10 mL of Qiagen’s buffer AE was used as the reference.
Quantitative PCR. The quantitative PCR reaction used primers, a generous gift from Dr. Rob DeSalle, that would amplify a ~800 bp fragment of the Drosophila melanogaster mtDNA cytochrome oxidase II region. The upstream primer used was named CoIIa and its sequence was 5’-GTTTAAGAGACCAGTACTTG-3’. The downstream primer was named CoIIb, sequence 5’-ATGGCAGATTAGTGCAATGG-3’.
A total of 35 mL of reaction were prepared using the reagents from a TaKaRa PCR Amplification Kit (TaKaRa Shuzo Co.). 1 mL of the DNA elution from each extraction was used, in 0.86 X PCR buffer (8.6 mM Tris-HCL, 43.0 mM KCL, 1.3 mM MgCl2), 214 mM each dNTP, 0.29 mM CoIIa, 0.29 mM CoIIb, and 0.88 U Taq DNA polymerase. The reactions were carried out on a Perkin-Elmer 9600 GeneAmp PCR System. The reaction conditions were as follows: 35 cycles of denaturing, annealing, and extension (94.0°C for 30 s, 60.0°C for 30 s, 72°C for 60 s), followed by storage at 4°C. The PCR products were stored at 4°C until agarose gel quantification.
Two PCR reactions were performed on each DNA extraction.
PCR Product Quantification. The PCR products were quantified on a 1.2% caterpillar (50-well) agarose/TBE gel. Five to six wells on each gel were loaded with varying amounts of Hi-Lo DNA Marker (Bionexus Inc): from 0.25 mg – 1.5 mg on each gel as a standard by which to quantify the amplified product. The same Hi-Lo solution was used as the standards in each gel, and the Hi-Lo solution was vortexed prior to each use in order to reduce variation in the quantification procedure. The gels were electrophoresed for ~270 min at 70V or overnight at 20V. After electrophoresis was complete, the gels were stained in 0.5 mg/mL ethidium bromide/TBE solution for 60 min, and destained in distilled water for 60 min. In the control and ethanol-preserved samples, a 12-well 1.2% agarose minigel was used to quantify the PCR products.
The gels were placed on a UV transilluminator and were scanned into
a computer using a Kodak DC290 camera, with a 3.5 s exposure time. The amount of CoII strand amplified in
each reaction mixture was then quantified using the Kodak 1-D Image Analysis
software. One of the Hi-Lo standards
was used as an experimental lane in order to test the accuracy of the quantification
procedure. The outlying bands
in the mass curve were ignored such that the R2 value for the curve
was closest to one and the curve provided the most accurate in quantifying
the known mass of DNA in the “experimental” standard.
Data Analysis
For each PCR reaction, the value for the mass of the PCR product (as measured by computer densitometry) was divided by the mass of template DNA used in the PCR reaction (as measured by spectrophotometry) to give the amplification fraction (Af). The Af is an indicator of the relative quality of the DNA, according to the linear relationship between the amount of template DNA used in the reaction and the amount of amplified product. If the template DNA is of high quality, a large amount of amplified product will be derived in the PCR reaction from a particular amount of template used, corresponding to a relatively high Af.
The statistical
test (Mann-Whitney Rank Sum) was performed using the Minitab data analysis
software to compare the means of the two PCR reactions for each protocol,
or to compare the results from one PCR reaction if enough data was not available. In Table 1, those samples that do not
include a standard deviation in addition to the amplification fraction were
calculated from a sample size of one; those samples that do include a standard
deviation were calculated from a sample size of two.
Results:
Table 1 presents
the complete results of the amplification fraction (Af) data for
samples preserved in each cryopreservation protocol, as well as the results
of the amplification fraction for the control samples.
Control Samples and Cryopreserved
Samples:
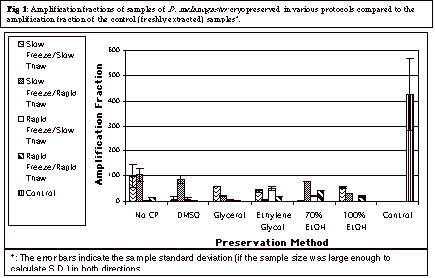
The control samples showed an Af of 425.48 ± 143.55, a number that was found to be significantly higher (Figure 1) than the set of the Afs of the cryopreserved samples (p < 0.01). The higher Af indicates significantly less damage (Chen et al., 1999; Ploskonosova et al., 1999; Ayala-Torres et al., 2000) in the DNA extracted from the fresh samples as compared to the DNA extracted from the cryopreserved samples .
|
Table 1. Amplification fractionsa
of samples of D. melanogaster cryopreserved using various protocols
as compared to the amplification fraction of the control samples.b
a: The amplification
fraction is a relative measure of the quality of the DNA in that it
signifies the mass of the amplified product (ng) per nanogram of template
DNA used in the reaction. Higher
quality DNA will have a higher amplification fraction. |
Rapid Freezing in Comparison
to Slow Freezing:
The mean Af (data not shown) for samples
preserved in all “six” cryoprotectants (the preservation of “No
cryoprotectant” is counted as a cryoprotectant) through the slow freeze
/ slow thaw protocol was found to be significantly higher than the mean Af
for samples similarly preserved in the rapid freeze / slow thaw protocol (p
< 0.05, Mann-Whitney Rank Sum Test). Additionally, the mean Af of
samples preserved in all “six” cryoprotectants in the slow freeze
/ rapid
thaw was significantly higher (p < 0.05) than the mean Af of
the samples subjected to the rapid freeze / rapid thaw protocol. These data are visually shown in Figure
2.
Optimal Cryopreservation Protocol:
The thawing protocol
was observed to make no difference in the Af (p > 0.6 in Mann-Whitney
Rank Sum test comparing the mean Afs of each thawing protocol for
all cryoprotectants in both the rapid freeze and the slow freeze protocol).
Therefore, it was decided to focus on which particular cryoprotectant
under which freezing method gave the best results.
Slow freezing using no cryoprotectant was found to have the highest
mean Af (p < 0.05 comparing to the set of Afs for
all cryoprotectant and freezing methods).
 |
No amplification was observed in any of the quantitative PCR reactions from the four D. pseudoobscura used in the study (two PCR reactions per single-fly extraction).
 |
The data in this report indicate genetic damage by cryopreservation, in patterns consistent with known data on the effects of cryopreservation (Bouquet et al., 1993; Shaw et al., 1991) . The significant increase in amplification fractions (see Methods) and, therefore, quality of DNA, from the control D. melanogaster samples to the cryopreserved samples indicate that cryopreservation does indeed damage DNA. Much of the previous data on mutation as a result of cryopreservation was done using tests that would indicate severe mutations and further effects of cryopreservation in vivo (Ross et al., 1990; Houle et al., 1997) . However, in the case of quantitative PCR, where a single base adduct can result in the cessation of reaction for a particular template DNA strand, the test is much more sensitive to slight injuries to DNA.
The finding that DNA is significantly damaged by cryopreservation can have implications for any type of genomic research that uses enzymatic amplification, including phylogenetic research. However, for most present genomic techniques, cryopreservation seems to be a valid option for long-term storage of tissues, especially when used for sequencing, which requires that only a short strand be used in the PCR reaction. Since storage at cryogenic temperatures essentially stops any further chemical reactions (Grout and Morris 1987) , it is likely that long-term cryopreservation would be a suitable method for storing tissues for molecular studies using most present techniques, as an adequate amount of PCR product from a <1 kb sequence can clearly be derived from cryopreserved samples. The same cannot be said for ethanol-preserved tissues. In the PCR test using 70% ethanol-preserved sample D. pseudoobscura, no amplification was observed whatsoever. The same CoII primers used in this study are designed for a highly conserved region and have been known to amplify in D. pseudoobscura before (Liu and Beckenbach 1992) , so primer incompatibility should not have been a problem. The quality of DNA suggested by the tests on the ethanol-preserved D. pseudoobscura samples cannot be directly compared to the measured DNA quality in the cryopreserved D. melanogaster samples, due to serious differences in experimental protocol between the two types of samples. However, the failure to amplify in D. pseudoobscura suggests that ethanol storage may not be the optimal preservation method for long-term molecular studies, consistent with past reports (Barnes et al., 2000) .
Although there were few identifiable patterns in the data for the measured Af for each cryoprotectant in each freeze/thaw protocol, the patterns that result are consistent with the results from previous studies on cryopreservation and cryoprotectants (Bouquet et al., 1993; Shaw et al., 1991) .
The nature of the freezing protocol has been found, in past cryopreservation studies, to be integral in assuring cell survival. In the absence of a vitrification freezing protocol, which requires careful control of cryoprotectant concentration, freezing rate and thawing rate, slow freezing has been found to generally increase post-thaw viability and reduce gross chromosomal aberrations (Kasai et al., 1981; Grout and Morris 1987; Shaw et al., 1991; Stanic et al., 2000) . The data from this experiment further iterates that slow freezing is superior in its ability to preserve DNA.
Cryoprotectants are usually used to protect cells or reproductive tissue from the stresses of freezing, although they are known to be toxic, and in some cases, genotoxic (Watson and Holt 2001) . DMSO is known to be genotoxic (Kapp and Eventoff 1980; Hakura et al., 1993) . There is conflicting data on whether or not the following chemicals (used as cryoprotectants in this experiment) are genotoxic: ethanol, glycerol, and ethylene glycol (McCann et al., 1975; Pfeiffer and Dunkelberg 1980; Tuite et al., 1981; Bariliak and Kozachuk 1985; Obe and Anderson 1987; Zeiger et al., 1987; Doolittle et al., 1988; Tateno et al., 1998; Blasiak et al., 2000) . If these cryoprotectants are indeed genotoxic, the fact that the D. melanogaster samples subjected to the slow freeze protocol were equilibrated an hour longer in cryoprotectant before freezing (in accordance with established protocols (Shaw et al., 1991) ) may have allowed the cryoprotectants to further damage the DNA. This would be consistent with the result that no cryoprotectant had a higher Af and, therefore, higher quality DNA. However, this is simply a conjecture, as the difference could be due to the fact that no serum was used in the cryoprotectant medium, or that the action of the surrounding solution caused extra/intracellular ice nucleation or, finally, that cell lysis occurred due to osmotic pressures (Karow and Critser 1997) .
There are many possibilities for future research on the utility of cryopreserving tissues for molecular studies. The quality of mRNA strands or long strands (2 – 40 kb) of DNA isolated from cryopreserved tissues is ambiguously defined, as these nucleic acids are more sensitive to chemical and physical stresses. The Taq used in PCR reactions is extremely temperamental, although techniques have recently been invented to amplify longer strands of DNA (LA-PCR), using proofreading enzymes such as Tfu. The long PCR protocol has many applications that would make the preservation of high molecular weight DNA in tissues a worthwhile goal: whole-genome mapping, analyzing homologous recombination events, and amplifying whole cDNAs (Cohen 1994) . Still, if there are too many adducts in these strands, the long PCR protocol will fail to synthesize strands of high molecular weight DNA. Therefore, some future research pathways might be: using a more sensitive quantitative PCR test using the long PCR protocol to amplify a 10-20 kb strand of DNA (Ayala-Torres et al., 2000) to determine the effects of cryopreservation on genomic as well as mitochondrial DNA, or to additionally determine the effects of cryopreservation on tissues from other invertebrate and vertebrate species. In addition, it may be worthwhile to perform this particular experiment again using more sensitive equipment to quantify the mass of the PCR product. Tests were done using the Kodak 1D software to quantify the bands of the Hi-Lo standards and consistent departures of ~15% from the known mass were found. The Kodak system was sensitive enough to find that slow freezing was superior to rapid freezing as well as find an optimal freezing procedure. However, if the experiment was to be performed again using tools more suitable for qualitative analysis, namely a better DNA stain (i.e., Sybr-Green), and a more accurate PCR product quantification system (i.e., Amersham Pharmacia’s Typhoon Phosphoimager), the results should have greater statistical significance and less measured error.
References: Ayala-Torres, S., Y. Chen, et al., 2000, Methods 22(2): 135-147; Bariliak, I.R., and S.I. Kozachuk 1985, Tsitol. Genet. 19(6): 436-442; Barnes, I., J. Holton, et al., 2000, J. Pathol. 192(4): 554-559; Balpaeme, K., K. Cooreman, et al., 1998, Mutat. Res. 415(3): 167-184; Blasiak, J., A. Trzeciak, et al., 2000, Toxicol. In Vitro 14(4): 287-295; Bouquet, M., J. Selva, et al., 1993, Biol. Reprod. 49(4): 764-769; Chen, X., C. Cullinane, et al., 1999, Mutat. Res. 445(1): 45-54; Cohen, J., 1994, Science 263(5153): 1564-1565; Dick, M., D.M. Bridge, et al., 1993, Methods Enzymol. 224: 51-65; Dillon, N., A.D. Austin, et al., 1996, Insect Mol. Biol. 5(1): 21-24; Doolittle, D.J., D.A. Lee, et al., 1988, Food Chem. Toxicol. 26(7): 631-635; Duru, N.K., M.S. Morshedi, et al., 2001, J. Androl. 22(4): 646-651; Fisher, R.L., S.J. Hasal, et al., 1996, Cryobiology 33(1): 163-171; Flournoy, L.E., R.P. Adams, et al., 1996, Biotechniques 20(4): 657-660; Grout, B.W.W.,and G.J. Morris 1987, The Effects of Low Temperatures on Biological Systems; Hakura, A., H. Mochida, et al., 1993, Mutat. Res. 303(3): 127-133; Honda, S., A. Weigel, et al., 2001, Biochem. Biophys. Res. Comm. 282(2): 493-498; Houle, D., A.S. Kondrashov, et al., 1997, Genet. Res. 69(3): 209-213; Ingham, E., J.B. Matthews, et al., 1993, Cryobiol. 30(5): 443-458; Kapp, R.W., Jr., and B.E. Eventoff 1980, Teratog. Carcinog. Mutagen. 1(2): 141-145; Karrow, A.M., and J.K. Critser 1997, Reproductive Tissue Banking: Scientific Principles; Kasai, M., K. Niwa, et al., 1981, J. Reprod. Fertil. 63(1): 175-180; Liu, H., and A.T. Beckenbach 1992, Mol. Phylogenet. Evol. 1(1): 41-52; McCann, J., E. Choi, et al., 1975, Proc. Natl. Acad. Sci. USA 72(12): 5135-5139; Obe, G., and D. Anderson 1987, Mutat. Res. 186(3): 177-200; Parkening, T.A., Y. Tsunoda, et al., 1976, J. Exp. Zool. 197(3): 369-374; Pfeiffer, E.H., H. Dunkelberg 1980, Food Cosmet. Toxicol. 18(2): 115-118; Ploskonosova, I.I., V.I. Baranov, et al., 1999, Mutat. Res. 434(2): 109-117; Ross, K.S., N.E. Haites, et al., 1990, J. Med. Genet. 27(9): 569-570; Shaw, J.M., I. Kola, et al., 1991, J. Reprod. Fertil. 91(1): 9-18; Stanic, P., M. Tandara, et al., 2000, Eur. J. Obstet. Gynecol. Reprod. Biol. 91(1): 65-70; Steele, E.K., N. McClure, et al., 2000, Fertil. Steril. 74(3): 450-453; Tateno, H., T. Wakayama, et al., 1998, Zygote 6(3): 233-238; Tuite, M.F., C.R. Mundy, et al., 1981, Genetics 98(4): 691-711; Watson, P.F., and W.V. Holt 2001, Cryobanking the Genetic Resource: Wildlife Conservation for the Future?; Zeiger, E., B. Anderson, et al., 1987, Environ. Mutagen. 9(suppl. 9): 1-109.