
Bertucci, Lisa A., and Mohamed A.F. Noor. 2001. Single fly total RNA preparations for RT-PCR.Dros. Inf. Serv. 84: 166-168.
|
|
|
|||
|
|
||||
Single fly total RNA preparations for RT-PCR.
Bertucci, Lisa A., and Mohamed A.F. Noor. Department of Biological Sciences, Louisiana State University, Baton Rouge, LA, 70803, USA.
Though several means of isolating DNA from a single fly have already
been documented (e.g., Gloor and Engels, 1992), we were unable to find a
published method of total RNA isolation from a single fly. Total RNA from a single fly can be used
for quantitative trait locus (QTL) mapping of expression profiles by RT-PCR.
Such RNA would need to be free of contaminating genomic DNA.
Here, we present a variant of the Ambion RNAwiz protocol that we have
tested on both single Drosophila simulans and single D. mauritiana,
and surely could be applied to single D. melanogaster. The protocol
takes just over two hours, though multiple samples can be prepared simultaneously,
and each preparation yields at least enough RNA for one or two RT-PCR reactions.
Preparation Protocol:
Required Supplies:
RNAwiz (Ambion Cat. #9736)
DNA-free (Ambion Cat. #1906)
GlycoBlue (Ambion Cat. #9515)
Chloroform (no isoamyl alcohol)
Isopropanol
75% ethanol (cold: -20ºC)
RNase-free water
Microcentrifuge pestle + tube (0.5ml preferred) (Fisher Scientific Cat. #K769520-0590)
Microcentrifuge
(maximum speed 13,000 RPM)
(1) Place one anesthetized fly into a microcentrifuge tube containing 50ml RNAwiz and homogenize for 1-2 minutes. Incubate for 5 minutes at room temperature.
(2) Add 10ml chloroform to homogenate. Shake tube vigorously for 20 seconds, and then allow the mixture to incubate for 10 minutes at room temperature.
(3) Centrifuge at maximum speed for 15 minutes at 4ºC.
(4) Transfer approximately 5ml of the top aqueous phase into a new tube, being careful not to disturb the interphase. Add 25ml of water, 0.5ml GlycoBlue, and 50ml isopropanol. Mix gently after each addition. Incubate for 10 minutes at room temperature. Then, centrifuge at maximum speed for 15 minutes at 4ºC. Discard the supernatant.
(5) Wash the pellet with 50ml of 75% cold ethanol by pipetting up and down to loosen the pellet from the tube wall. Centrifuge at maximum speed for 5 minutes at 4ºC. Discard the supernatant.
(6) Allow the RNA pellet to air dry for 10 minutes.
(7) Resuspend the RNA pellet in 10ml of water. Add 1ml of 10´ DNase I Buffer and mix. To denature any remaining DNA, add 0.5ml DNase I (1 unit), mix, and incubate for 30 minutes at 37ºC.
(8) Add 3ml of vortexed DNase Inactivation Reagent to the tube containing the RNA and mix well. Incubate for 2 minutes at room temperature.
(9) Centrifuge at maximum speed for 1 minute to pellet the inactivation reagent. Transfer 10ml of the supernatant to a new tube to be used in reverse transcription.
RNA Concentration and Purity
Data
We evaluated the yield of thirteen single fly RNA preparations. All flies used were males of either Drosophila simulans or D. mauritiana. Our RNA preparations yielded 920 ± 80ng (mean ± s.e.) total RNA, as determined by UV spectrophotometry. The average ratio of OD260 to OD280 values was 1.68 ± 0.05 (mean ± s.e.), which is slightly less than the ideal of 1.90 and thus indicates incomplete purity and some possible protein contamination in the isolated RNA samples. However, the OD320 value was consistently zero, indicating a lack of particulate matter.
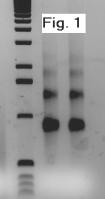
The quality of the RNA from this preparation was tested for degradation
by running two samples on a 1% TBE agarose gel.
Clear ribosomal RNA bands were visualized from both samples around
800 and 1300bp, indicating no significant degradation in the isolated RNA
(see Figure 1). To test the usability
of the samples in RT-PCR, separate reverse transcription (RT) and polymerase
chain reactions (PCRs) were done on two of the RNA samples. We selected primers flanking an intron of the schnurri gene for this test to evaluate also whether genomic
DNA contamination remained. Half
of the sample was first assayed in a spectrophotometer to determine its concentration.
The remaining halves of the samples, one bearing 320ng and one bearing
480ng RNA, were used inreverse transcription reactions (5mM MgCl2,
50mM KCl, 10mM Tris-HCl, 1mM dNTPs, 20U RNasin, 20U reverse transcriptase,
2.5 mM primer,
RNA sample). The resulting cDNA
was then used in a PCR reaction bearing 2ml of the RT rea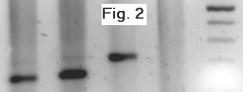 ction
(PCR reaction conditions 10mM Tris-HCl, 50mM KCl, 2.2 mM MgCl2,
0.2mM dNTPs, 1mM
of each of two primers, 1U Taq polymerase). A D. mauritiana genomic
DNA sample and a negative control were also amplified from the same master
mix. PCRs were visualized on
a 1% TBE agarose gel. Clear amplification
was observed from the RT samples at the expected smaller size than the genomic
DNA sample (see Figure 2), and no amplification was observed from the negative
control.
ction
(PCR reaction conditions 10mM Tris-HCl, 50mM KCl, 2.2 mM MgCl2,
0.2mM dNTPs, 1mM
of each of two primers, 1U Taq polymerase). A D. mauritiana genomic
DNA sample and a negative control were also amplified from the same master
mix. PCRs were visualized on
a 1% TBE agarose gel. Clear amplification
was observed from the RT samples at the expected smaller size than the genomic
DNA sample (see Figure 2), and no amplification was observed from the negative
control.
References: Gloor, G., and W. Engels 1992, Dros. Inf. Serv. 71: 148-149.