Cabral, H., M.T. Ruiz, C.M.A. Carareto and G.O. Bonilla-Rodriguez. 2000. A plant proteinase, extracted from Bromelia fastuosa, as an alternative to proteinase K for DNA extraction. Dros. Inf. Serv. 83:178-185.
|
|
|
|||
A plant proteinase, extracted from Bromelia fastuosa, as an alternative to proteinase K for DNA extraction.
Cabral1, H., M.T.
Ruiz1, C.M.A. Carareto2 and G.O. Bonilla-Rodriguez3. 1Graduate student, 2Departamento de Biologia, 3Departamento de
Química e Geociências, Ibilce-Unesp, Rua Cristovão Colombo
2265, São José do Rio Preto SP, Brasil. Correspondence: Gustavo
O. Bonilla Rodriguez. Departamento de Química e Geociências,
IBILCE-UNESP, Rua Cristóvão Colombo 2265, CEP 15054-000, São
José do Rio Preto SP, Brasil. bonilla@qeg.ibilce.unesp.br
Abstract: We report the use of brofasin, a proteolytic enzyme extracted
from the fruits of Bromelia fastuosa, as a protective agent in DNA extraction protocols.
Brofasin at concentrations varying between 236 units/µg to 2,372 units/µg
was used to extract DNA of human leukocytes and Drosophila melanogaster
tissues. At brofasin concentration between 1,186 and 2,372 units/µg
the resulting DNA concentrations are similar to those obtained by treatment with proteinase K. The good yield of extracted
DNA and its intactness and availability for further analysis, verified by
PCR and restriction enzyme digestion reactions, suggest the use of this enzyme
as an alternative to proteinase K.
Introduction
Proteinases are hydrolytic enzymes used by industrial processing to
transform proteins in shorter peptides for several purposes (Flynn, 1975).
These enzymes act by hydrolizing peptide bonds and exhibit a wide range of
cleavage preferences. Extraction and purification of several plant proteinases
can be relatively easy and can be done with low-cost procedures. That is the
example of papain, used in meat tenderization and beverage clarification (Flynn,
1975; Castro, 1981; Salunke and Desai, 1984; Poulter and Caygill, 1985).
Proteinases can be also used as protective agents during DNA extraction,
by inactivating endogenous nucleases. Proteinase K, a Ca2+-dependent
enzyme extracted from Tritirachium album (Ebeling et al., 1974) is largely used with that purpose, remaining
substantially active during incubation above 50ºC in SDS-containing buffers (Blind
and Stafford, 1976; Krusius and Finne, 1982).
Recently Genelhu et al. (1998) isolated by gel filtration
and ion-exchange chromatography a 22-24 kD plant cysteine-proteinase from
Carica candamarcensis which
was named as E6870. The authors proposed the use of this enzyme as a good
option as protective agent during isolation of bacterial genomic DNA.
The present work is part of a research involving the purification and
characterization of proteolytic enzymes from the fruits of Bromelia fastuosa (known as gravatá). Preliminary
studies performed with enzyme inhibitors (Mateus et al., 2000; Cabral et al., 2000; Romanelli et al., 1994) classified this enzyme, named
brofasin, as a cysteine-proteinase, a class of enzymes having a cysteine and
a histidine residues in the active site (Dunn, 1996).
As low-cost alternative products are
very welcome in molecular biology procedures, we report
results obtained with brofasin which illustrate its use as a protective agent
in DNA extraction protocols from human leukocytes and Drosophila melanogaster
tissues in comparison with proteinase K.
Material and Methods
All purification procedures concerning brofasin were carried out at
low temperature (0-4ºC). Purification
of the proteinase started with
salt precipitation using ammonium sulphate (AS) of the extract obtained from
the pulp of unripe fruits. The precipitation procedure started with 50% AS,
decreasing progressively to 30%. The supernatant, at this concentration, exhibited
the highest level of proteolytic activity. After dialysis against buffer A
(70 mM acetate buffer pH 3.6) the supernatant was applied to a 5 ´ 11 cm BioRad column containing CM-Sepharose equilibrated
with buffer A. The elution was done by salt gradient against buffer A containing
2 M sodium chloride. The fractions containing proteolytic activity were pooled,
concentrated by centrifugation on Amicon centriprep-10 and submitted to gel-filtration.
This operation was performed using a 1.5 ´ 70 cm BioRad column and Sephadex G-50 (Sigma).
The enzyme containing fractions were applied subsequently to isoelectric focusing
performed in a Rotofor Cell (BioRad) using an EPS 3501 power supply (Pharmacia) and cooled with a refrigerated
circulating water bath (Lauda WK-500).
Proteolytic activity was measured using 1% casein (Sigma), stopping
the reaction with 10% trichloroacetic acid (TCA). Every test had its own blank.
The increase of absorbance at 280 nm in the supernatant after centrifugation
is proportional to soluble peptides that remain in solution (Sarath, 1996).
A unit of enzyme activity was defined as the amount of enzyme required to
produce release one µmol of Tyrosine per minute in the essay. To relate
the absorbance reading at 280 nm with Tyrosine concentration we used an extinction
coefficient of 0.005 ml µg-1 cm-1 conditions (Moyano-López et al., 1999).
Suitability
of brofasin was tested for genomic DNA extraction from insect and human cells.
For insects we tested two protocols
for DNA extraction from Drosophila melanogaster, which differ only in terms of the buffer. The
first protocol used an extraction buffer containing urea (7M urea, 350 mM
NaCl, 20 mM EDTA, 1% n-lauryl sarcosine and 0.5 % SDS). The second method
used a different buffer, without urea (10 mM Tris-HCl pH 8.0, 10 mM EDTA,
350 mM NaCl, 0.5 % SDS). For both experiments we used six experimental groups,
each constituted by 15 specimens, which were squashed with the extraction
buffers. The first two groups were the positive control (group 1), with 400
units/µg of proteinase K, and the negative control (group 2), without
any enzyme. The brofasin containing groups (3 to 6) contained, respectively,
2,372 units/µg, 1,185 units/µg,
474 units/µg and 236 units/µg of brofasin.
The eppendorf tubes of each experimental group were incubated at 50ºC
for 20 minutes, followed by a standard phenol/chloroform/isoamyl alcohol extraction
and sodium acetate/absolute ethanol precipitation (Sambrook et al., 1989). After washing with 70% ethanol,
the dried pellet was dissolved in 20 ml TE (Tris-EDTA)
and 10 ml of this solution was electrophoresed
on a 1% agarose gel in TAE buffer containing 0.5µg of ethidium bromide.
A sample of this DNA was also spectrophotometrically quantified. To check for possible adverse effects on DNA, the intactness of DNA resulting from this extraction was evaluated by applying the PCR technique. The PCR reaction was carried out by standard methods using oligonucleotide primers that flank a 764 bp fragment of P transposable element, a middle repetitive sequence DNA of D. melanogaster genomes (primer 829: 5´-AACATAAGGTGGTCCCGTCG 3´ is complementary to nucleotides 12 to 31 of a complete P element and the primer 830: 5´-CGACTGGGAAAGGAAATCC 3´ is complementary to nucleotides 757 to 776). Temperature cycling was performed with the following profile: 92°C for 1 min; 50°C for 1 min; 72°C for 1 min, for a total of 30 cycles.
|
|
We also performed an additional
evaluation of DNA intactness carrying out restriction enzyme digestion with
endonuclease HindIII,
performed according to manufacturers recommendation.
In a second group of experiments we
evaluated the performance of brofasin to obtain human genomic DNA. Lymphocytes
from collected blood samples were isolated using the Ficoll-Histopaque procedure.
DNA was isolated from lymphocytes by the salting out extraction procedure
(Abdel-Rahman et al., 1994). To each eppendorf tube containing the isolated lymphocytes were
added the lysis buffer (10 mM Tris-HCl, 400 mM NaCl, 2 mM EDTA) and 28.8 units/µg
of RNase A, constituting three experimental groups: the negative control group
(1), without any proteinase, the positive control group (2), to which was
added 20 units/µg of proteinase K, and the brofasin group (3), to which
was added 65.3 units/µg of
the enzyme. Precipitation was
performed with sodium chloride/absolute ethanol. After washing with 70% ethanol,
the dried pellet was dissolved in 300 ml H2O
and 10 ml of this solution was electrophoresed
on a 1% agarose gel in TAE buffer containing 0.5µg of ethidium bromide.
DNA concentration was measured
spectrophotometrically.
The viability of the resulting DNA was checked by amplification of
a sequence of the CYP2E1 gene (locus 1370 - 1349: 5´-CCA GTC GAG TCT
ACA TTG TCA and locus 999-978: 5´- TTC ATT CTG TCT AAC TGG) by the PCR
reaction consisting of 25 cycles of denaturation at 95ºC (1 minute), annealing at 55ºC (1 minute) and extension at
72ºC (1 minute). The DNA intactness was also checked by a restriction
enzyme digestion with endonuclease EcoRI performed according to manufacturers recommendation.
The extracted DNA from Drosophila
and humans, their products of amplification and digested fragments were separated
in a 1% agarose gel in TAE buffer containing 0.5µg of ethidium bromide.
Results and Discussion
Table 1 shows the concentrations of
genomic DNA of D. melanogaster obtained with both kinds of extraction buffers. In those groups where
extraction buffer containing urea was used, higher amounts of DNA were recovered. Group 3 (2,372 units/µg of brofasin)
yield the higher amount of DNA
(349.5 ng/µL), even higher than the proteinase K group (212.3 ng/µL).
The amount in groups 1, 2 and 4 were similar and groups 5 and 6 showed a significant
decrease in DNA yield. Without urea the concentration of 236 units/µg
of brofasin (group 6) was shown to be inadequate for DNA extraction. Concerning
the tests using extraction buffers without urea, the amount of recovered DNA
was significantly inferior compared to the urea buffer, in all groups; in
the three experimental groups containing brofasin (3, 4 and 5), DNA concentration
varied between 111.1 and 174.5 ng/µL, being higher than those obtained
with proteinase K (85.6 ng/µL).
Samples of these genomic DNA (10 µL)
that was run in 1% agarose gels are shown in Figures 1 A (DNA extracted with urea) and 2A (DNA
extracted without urea).
More important than the amount of DNA is the quality and its availability
for ulterior analysis. PCR amplification of P sequences produced the expected fragment with
764 bp, for almost all the concentrations of brofasin and the positive control
as well (Figures 1B and 2B). The only failure on obtaining amplification occurred
for group 6 in the absence of urea (Figure 2B-6), probably due to the low
template concentration. The DNA produced in the negative control (extraction
without proteinase K) also did not show amplification (Figures 1B and 2B),
independently of the buffer used.
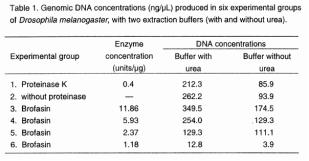
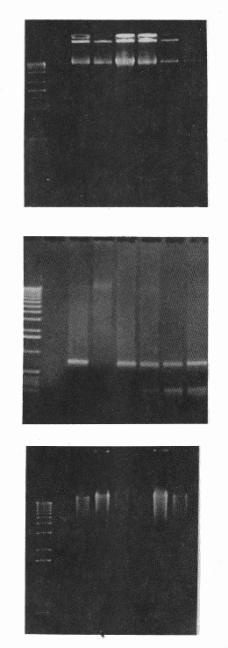 |
Figure 1 (A, top; B, middle; C, bottom). DNA from 15 D. melanogaster specimens which were squashed with extraction buffer containing urea (7M urea; 350 mM NaCl;
20 mM EDTA; 1% n-lauryl sarcosine; 0.5 % SDS) to which it was added proteinase
K, brofasin or no enzyme, followed by phenol extraction (phenol:
chlorophorm: isoamylalchool = 25:24:1). A: Genomic DNA extracted with different concentraions
of proteases, or without enzyme. Lanes: 1- with 0.4 units/µg of
proteinase K; 2 - without
any enzyme; 3- with 11.86
units/µg of brofasin; 4-
with 5.93 units/µg of brofasin; 5- with 2.37 units/µg of brofasin; 6- with 1.18 units/µg of brofasin;
B: Amplification of 764 bp of P-element transposon;
C: Genomic Hind
III digests. |
 |
| Figure 2 (A, left; B, right). DNA from 15 D. melanogaster
specimens which were squashed with extraction
buffer without urea (10mMTris-HCl pH 8,0; 10 mM EDTA; 350mM NaCl; 0.5 % SDS) to
which it was added proteinase K, brofasin or no enzyme, followed by phenol
extraction (phenol:chlorophorm:isoamylalchool = 25:24:1). A: Genomic DNA extracted with different concentraions
of proteases, or without enzyme. Lanes: 1- with 0.4 units/µg of proteinase K; 2 - any enzyme; 3- with 11.86 units/µg of brofasin;
4- with 5.93 units/µg of brofasin;
5. with 2.37 units/µg of brofasin;
6- with 1.18 units/µg of brofasin; B: Amplification of 764 bp of P-element transposon;
C: Genomic Hind III digests. |
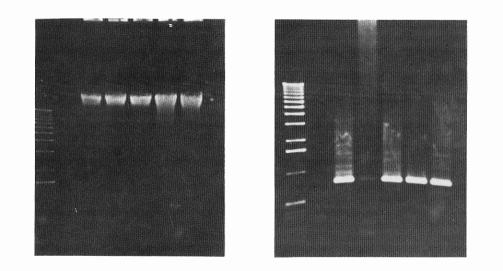
Human genomic DNA was obtained with the following concentrations: 309.3
ng/µL for the negative control, 462.6 ng/µL for the positive control
(proteinase K) and 498.6 ng/µL for the brofasin containing group (Figure
3). The effects of brofasin and proteinase K were quite similar. The DNA yield
in group 1, without proteinase, were high, however, note in Figure 3A that
the band corresponding to it shows a smear suggesting degraded DNA. The DNA
quality and its suitability for molecular techniques was verified by the PCR
reaction, where for all the samples, the sequence of the CYPE1 gene with about
400 bp was amplified. Digestion with restriction endonuclease EcoRI was equally suitable for the groups
containing proteinase K (2) and brofasin (3), but not for the negative control
(1).
Proteinase K is frequently used for DNA extraction due its efficiency
for digests of nuclei or whole cells and release of DNA for the action of
polymerases. It has the advantage of being stable on relatively high temperatures
(55 to 60ºC), being easily inactivated at 95ºC, an important characteristic
during PCR reaction.
Our results show that the enzyme brofasin is equally efficient for nuclease
inactivation during DNA extraction. The good yield of extracted DNA and the
intactness and availability of DNA resulting from the protective action of
brofasin for ulterior analysis, verified by PCR and restriction enzyme digestion
reactions, suggest the use of this enzyme as an alternative to proteinase
K for procedures involving DNA extraction.
 |
| Figure 3 (A, left; B, right). Human genomic DNA extracted from lymphocytes be the salting out extraction procedure, using as lysis buffer 10 mM Tris-HCl, 400 mM NaCl, 2 mM EDTA and 28.8 units/µg of RNase A, to which was added proetinase K, brofasin or no enzyme. A: Genomic DNA extracted with different concentrations of proteases, or without enzyme (a), and genomic DNA EcoRI digests (b): Lanes: 1- withou any enzyme; 2- with 20 units/µg of proteinase K; 3- with 65.3 units/µg of brofasin; B: Amplification of 400 bp of the CYP2E1 gene. |
Acknowledgments: This research was supported by
FAPESP (Fundação de Amparo à Pesquisa do Estado de São
Paulo) (No 1995/7192-2
and 1998/08734-1) and by a CAPES fellowship (Coordenação
de Aperfeiçoamento de Pessoal de Nível Superior) to H.C. and
M.T.R. We thank to N.D.T.C. Froes for providing the CYP primers and to A.R. Chinelato
and ARB Rossit for technical support.
References: Abdel-Rahman, S. Z., A. M. Nouraldeen,
and A. E. Ahmed 1994, J. Biochem. Toxicology, 9: 191-198; Blin, N., and D.W. Stafford 1976, Nucleic
Acids Research, 3: 2303‑2307; Cabral, H., R.P. Mateus, C.R. Ceron,
and G.O. Bonilla-Rodriguez 2000, in preparation; Castro, I.R. 1981, Natural Science Department Bureau Technology Journal,
23: 61‑66; Dunn, M.J.,
1989 In: Protein Purification Methods: A Pratical Approach. Publ. by IRL Press, NY, USA.; Ebeling, W., N. Hennrich, M. Kclockow,
H. Metz, H.D. Orth, and H. Lang 1974, Eur. J. Biochem. 47: 91‑97; Finne, J., and T. Krusius 1982, Methods
in Enzimology, 83: 269‑277; Flynn,G.
1975, In: Tropical Products, Institute Series. Ministry of Overseas Development,
London: 1‑52; Gehelhu,
M.S., M.S. Zanini, I.F. Veloso, A.M.D. Carneiro, M.T.P. Lopes, and C.E. Salas
1998, Brazilian Journal of Medical and Biological Research, 31: 1129‑1132;
Kawasaki, E.S., 1990, In: PCR
Protocols: A Guide to Methods and Applications, 166 pp.; Mateus, R.P., H. Cabral, G.O. Bonilla Rodriguez, and
C.R. Ceron 2000, Submitted to
Braz. J. Med. Biol. Res.; Moraes, M.G., C. Termignoni, and C.
Salas 1994, Plant Science, 102:11‑18; Moyano-López, F.J., I. Martínez-Díaz,
M. Díaz-López, and F.J. Alarcón-López 1999, Comp.
Biochem. Physiol. B 122:327-332; Poulter, N.H., and J.C. Caygill 1985, Tropical Sciences,
25: 123‑137; Romanelli,
P.F., E.L.M. Torres-Queiróz, and C.R. Ceron 1994, Bol. SBCTA 28: 120‑133; Salunkhe, D.K., and B.B. Desai
1984, In: Postharvest Biotechnology
of Fruits. (Fasman, G.D., ed.), vol. 2
CRC Press, Boca Raton: 3‑27;
Sambrook, J.
E.F. Fritisch, and T. Maniatis 1989, Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, New York; Sarath, G., R.S. de la Motte, and F.W.
Wagner 1996, In: Proteolytic Enzymes: A Practical Approach. (Beynon, R.J., and J.S. Bond, eds.), pp. 25‑55, IRL Press.