Matioli, Sergio Russo, and Flora Maria de Campos Fernandes-Matioli. 2001. A new pedagogical approach to teach phylogenetic reconstruction using distance data. Dros. Inf. Serv. 84:215-217.
|
|
|
|||
|
|
||||
A new pedagogical approach to teach phylogenetic reconstruction using distance data.
Matioli, Sergio Russo, and Flora Maria de Campos Fernandes-Matioli. Departamento de Biologia, Instituto de Biociências, Universidade de São Paulo. Correspondence to: Sergio Russo Matioli. Departamento de Biologia, Instituto de Biociências, USP, Caixa Postal 11.461, CEP 05422-970, São Paulo, Brazil. E-mail: srmatiol@ib.usp.br
Introduction
The phylogenetic reconstruction problem is a major issue in biology today. In every recent book on evolution and bioinformatics, this problem is carefully treated because there is a number of alternative methods that can be used with molecular data. The different methods are classified according to their principles, but there is no consensus in the classification. The distance methods, as traditionally labeled (Nei and Kumar, 2000), are also referred to as algorithmic methods (Swoford et al., 1996) and geometric methods (Russo et al., 2001). The distance methods are performed in two distinct steps. In the first step, pairwise distances are calculated upon the observed differences between two sequences. In this step, a model of evolution is considered to account for multiple substitution at each site. In the second step, the distance matrix is transformed in a tree. The goal is to accommodate the distances in a tree-like diagram. The transformation of distances into phylogenetic trees needs the concepts of additivity, ultrametricity, root and errors that are usually referred to triangle properties for teaching purposes We propose a new pedagogical approach to construct a tree diagram from a distance matrix.
Materials and Methods
|
Cut three nylon ropes of different
colors in ten pieces each with the suggested sizes presented in Table 1.
Attach a label with a single letter to each rope tip as indicated.
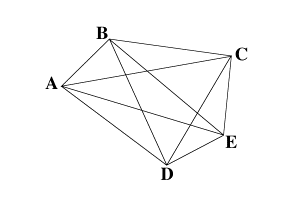
Three different demonstrations can be performed. We usually start the demonstration with the ultrametric set. Five students are asked to volunteer the demonstration. To each volunteer, a letter from A to E is assigned. The students are disposed in a circle. Four tips with the same letter as assigned to the student are given to him/her, forming a pattern as shown in Figure 1. The labels are aligned and the ropes tied with an adhesive tape just below the labels. The students are then informed of the geometric nature of the problem, where the distance matrix corresponds to distances between each pair of sequences or taxa.
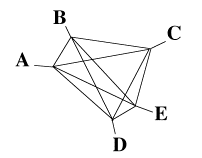
The "algorithm" employed here is purely mechanical. The problem given to the students is to construct a diagram with the shape of a phylogenetic tree by connecting the ropes in nodes where the ropes are perfectly aligned. The rope alignment may start from the tips, as shown in Figure 2. When the alignment of all the ropes set is achieved (by trial and error), the nodes are attached with adhesive tape. The teacher can, with the help of some students, show that there is a single point in the assembled ropes that is equidistant to all the tips. The students are informed that this reflects the property of ultrametricity, and, in this case, this point corresponds to the tree root.
|
The same procedure is applied to the additive set. In this case, a tree can be constructed, but it is impossible to find a point in the assembled ropes that has the same distance to the tips, showing that the property of ultrametricity no longer holds. The concept of tree root can also be discussed at this moment, as the use of an outgroup (the point that connect the tree to a known external sequence or taxon), the midpoint procedure (the midpoint of the largest distance), some sort of gravitation center, and so forth.
In the last demonstration, with the error set, it is even impossible to assemble the rope pieces co-linearly. The students are then asked how to solve the problem. The main issue, in this case, is how to distribute the errors (e.g., the use of elastic material for the ropes). This procedure would be analogous to those that are used with the computational algorithms (error evenly distributed, proportional to the lengths, and so forth).
If a computational facility is available to the students, they can use the matrix correspondent to the rope lengths in a computational program (e.g., the program neighbor from the PHYLIP package, Felsenstein, 1989. This package is recommended because it is freely available for the major computer platforms). In the case of the ultrametric set, both UPGMA and Neighbor-joining options will give the same results. With the additivity set, the results will be different according to the method, and so with the error set. In the last case, the tree distances will not correspond to the distance matrix. It is important to alert the students that the distance matrix should be analyzed for the properties of additivity and ultrametricity before a proper method can be employed and that UPGMA always produces an ultrametric tree, even if the distances in the matrix are not ultrametric.
|
Acknowledgments: The authors thank the students that have participated in the demonstrations performed during last few years in classes of "Evolutionary processes" and "Bioinformatics applied to molecular evolution and phylogenetics" carried out in University of São Paulo, Brasil.
References: Felsenstein, J., 1989, Cladistics 5: 164-166; Nei, M., and S. Kumar 2000, Molecular Evolution and Phylogenetics. Oxford Univ. Press, NY.; Russo, C.A.M., C.Y. Miyaki, and S.L. Pereira 2001, In: Biologia molecular e evolução (Matioli, S.R., ed.). Holos, R.P.; Swofford, D.L., G.J. Olsen, P.J. Waddell, and D.M. Hillis 1996, In: Molecular Systematics, 2nd Edition (Hillis, D.M., C. Moritz, and B.K. Mable, eds.). Sinauer Associates, Sunderland, M.A.